Chai-1: Neues KI-Modell übertrifft Google Deepminds AlphaFold bei Protein-Vorhersagen
Das KI-Start-up Chai Discovery präsentiert mit Chai-1 ein neues Modell zur Vorhersage komplexer Biomolekülstrukturen. Es übertrifft laut dem Unternehmen bestehende Methoden in vielen Bereichen und soll die Arzneimittelforschung beschleunigen.
Chai Discovery, ein KI-Unternehmen im Bereich der Arzneimittelforschung, hat ein neues KI-Modell namens Chai-1 entwickelt. Dieses Modell kann die dreidimensionale Struktur von Biomolekülen wie Proteinen und Nukleinsäuren mit hoher Genauigkeit vorhersagen.
Die Kenntnis der räumlichen Struktur von Biomolekülen ist entscheidend, um ihre Funktion und Wechselwirkungen zu verstehen. Dies bildet die Grundlage für die Entwicklung neuer Medikamente, die gezielt an bestimmte Moleküle im Körper binden sollen.
Chai-1 nutzt wie Deepminds AlphaFold 2 und AlphaFold 3 maschinelles Lernen, um aus den Sequenzen von Proteinen und anderen Biomolekülen deren dreidimensionale Struktur abzuleiten. Das Modell wurde mit einer großen Menge an Strukturdaten trainiert und kann nun auch für unbekannte Moleküle Vorhersagen treffen.
Laut den Entwicklern erzielt Chai-1 in verschiedenen Bereichen Spitzenleistungen:
Bei der Vorhersage von Protein-Ligand-Komplexen, also der Bindung kleiner Moleküle an Proteine, erreicht es eine Erfolgsrate von 77%. Dies entspricht dem aktuellen Bestwert des Modells AlphaFold 3. Bei der Vorhersage von Protein-Protein-Wechselwirkungen übertrifft Chai-1 mit einer Erfolgsrate von 75,1% das bisherige Spitzenmodell AlphaFold Multimer 2.3 (67,7%). Besonders gut schneidet Chai-1 bei der Vorhersage von Antikörper-Protein-Komplexen ab. Hier liegt die Erfolgsrate bei 52,9%, deutlich über AlphaFold Multimer 2.3 mit 38%.
Auch bei der Faltung einzelner Proteine zeigt Chai-1 sehr gute Leistungen und übertrifft knapp AlphaFold 2.3.
Chai-1 erzielt auch mit wenig Informationen gute Ergebnisse
Eine Besonderheit von Chai-1 ist, dass es auch ohne evolutionäre Sequenzinformationen (sogenannte Multiple Sequence Alignments) gute Vorhersagen treffen kann. In diesem "Single-Sequence-Modus" erzielt es bei Protein-Protein-Komplexen ähnlich gute Ergebnisse wie AlphaFold mit zusätzlichen Sequenzinformationen. Das ist insbesondere dort hilfreich, wo diese MSAs nicht vorliegen.
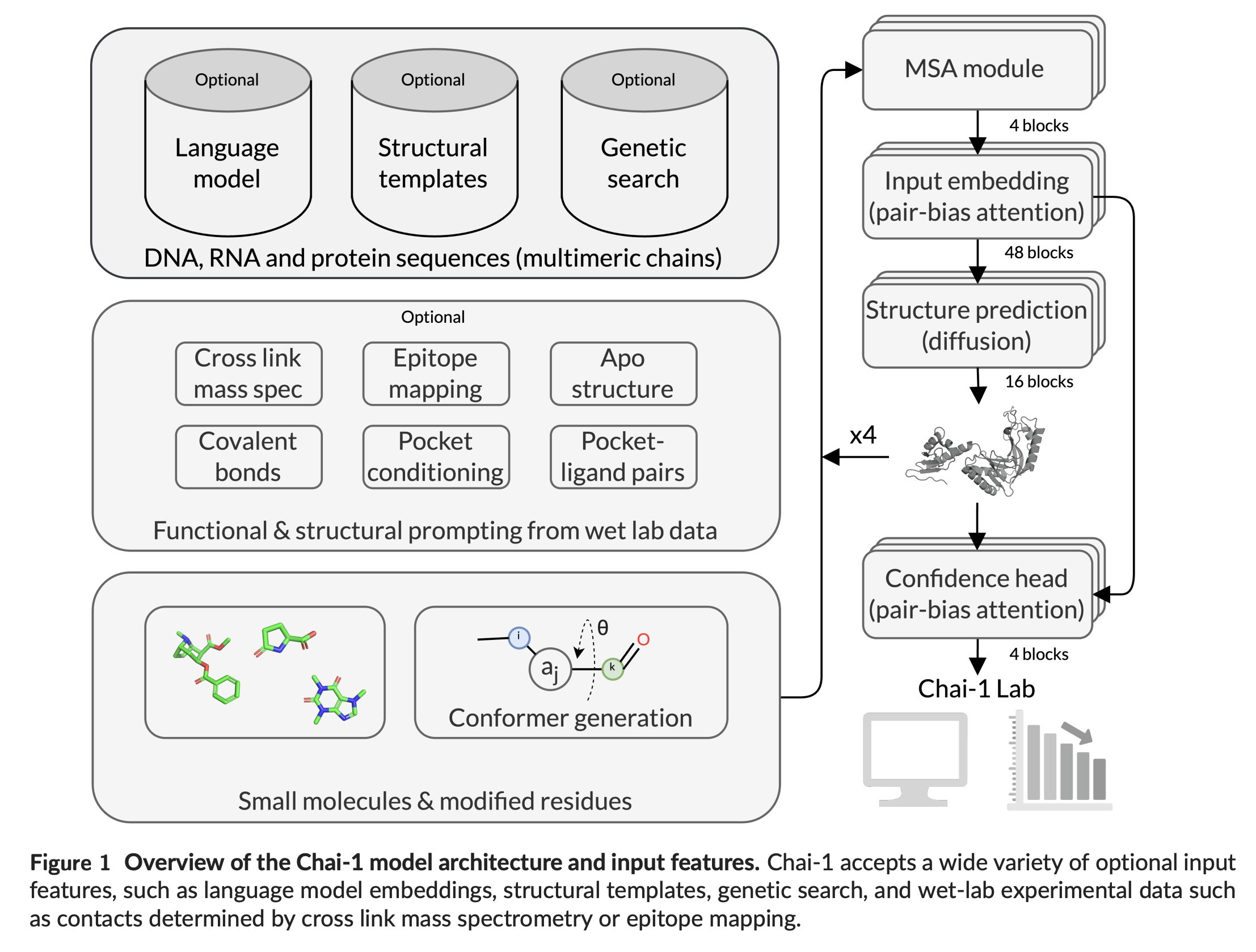
Ein weiterer Vorteil ist die Möglichkeit, experimentelle Daten als zusätzliche Informationen einzubeziehen. Wenn dem Modell beispielsweise Kontaktpunkte zwischen zwei Proteinen vorgegeben werden, verbessert dies die Vorhersagegenauigkeit erheblich.
Die Entwickler stellen die Gewichte und den Code des Modells für nicht-kommerzielle Zwecke zur Verfügung. Zudem bieten sie eine Web-Schnittstelle an, über die das Modell auch für kommerzielle Zwecke in der Arzneimittelforschung genutzt werden kann.
KI-News ohne Hype – von Menschen kuratiert
Mit dem THE‑DECODER‑Abo liest du werbefrei und wirst Teil unserer Community: Diskutiere im Kommentarsystem, erhalte unseren wöchentlichen KI‑Newsletter, 6× im Jahr den "KI Radar"‑Frontier‑Newsletter mit den neuesten Entwicklungen aus der Spitze der KI‑Forschung, bis zu 25 % Rabatt auf KI Pro‑Events und Zugriff auf das komplette Archiv der letzten zehn Jahre.
Jetzt abonnierenKI-News ohne Hype
Von Menschen kuratiert.
- Mehr als 16 % Rabatt.
- Lesen ohne Ablenkung – keine Google-Werbebanner.
- Zugang zum Kommentarsystem und Austausch mit der Community.
- Wöchentlicher KI-Newsletter.
- 6× jährlich: “KI Radar” – Deep-Dives zu den wichtigsten KI-Themen.
- Bis zu 25 % Rabatt auf KI Pro Online-Events.
- Zugang zum kompletten Archiv der letzten zehn Jahre.
- Die neuesten KI‑Infos von The Decoder – klar und auf den Punkt.